Chapter 5 - Command Line Tools for Genomic Data Science
quote?
This section will guide one through the process of ‘RNA-Seq’ or Transcriptome Analysis
In the study of Genomics there are three general questions which are particularly suited for
Typical Questions & Problems Regarding
1) Assembly
What genes are expressed in the sample?
What transcripts are expressed for each gene?
2) Quantification
What are the genes and transcripts expression levels?
3) Differential splicing and expression
How do expression levels and/or splicing patterns differ between two conditions (e.g., healthy vs disease tissue)?
5.1 Transcriptomic RNA-Seq Analysis
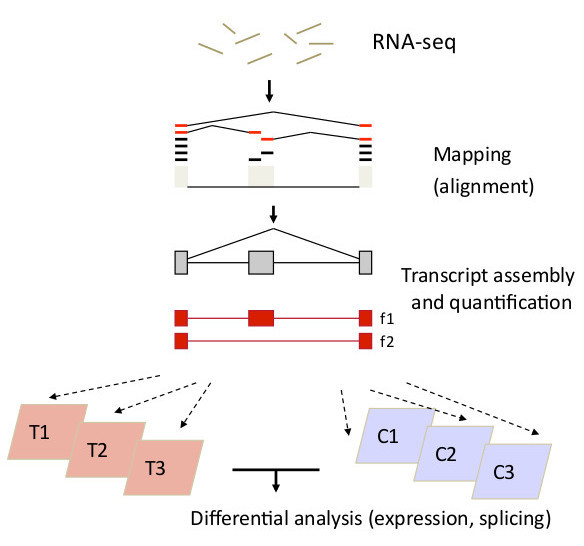
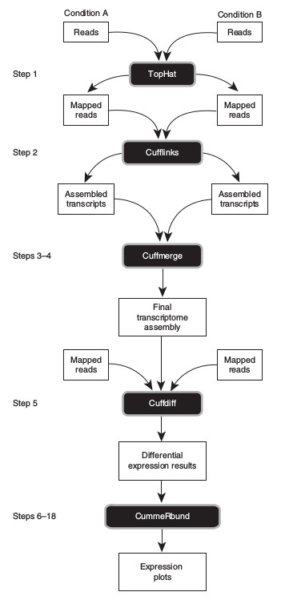
Trapnell RNA-Seq Flowchart
Transcript Assembly & Quantification
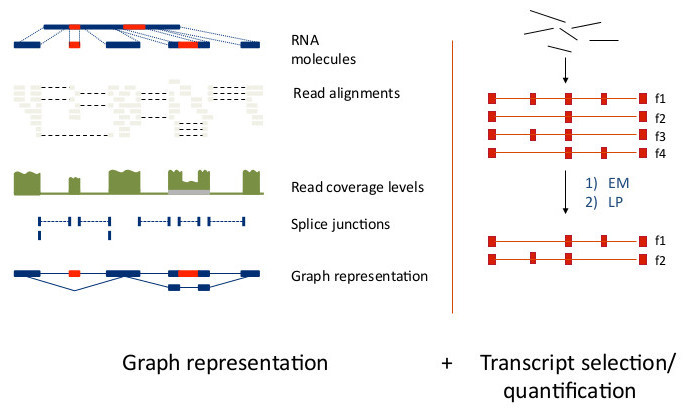
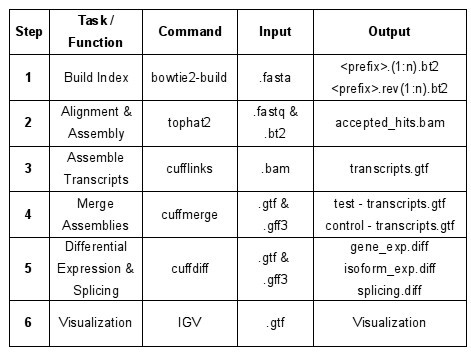
For this exercise this is RNA-seq analysis software
What you need:
Tuxedo suite - Trapnell et al., Nature Protocols 2012
- For a Very Good explanation of RNA-Seq see: The Griffith Lab, RNA-Seq Tutorial
Step 1: What is Tophat?
- TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions, D. Kim, et al, Genome Biology, 2013, 14:R36
- For even more information goto Tophat2.
It is always a good idea to READ OVER THE SOFTWARE before using it… Now, go to the command line:
1 # For version information:
2 tophat2 --version
3
4 # If you want the simple help print out try:
5 tophat2 >& tophat2.help
6 nano tophat2.help # nano is a very simple text editor
7
8 # Or view the manual:
9 man tophat2
For this experiment we will use a control set of paired end reads followed by a test set of paired end reads. Both samples will be in triplicate.
Now, let us inspect our data for read length and file integrity.
1 cd dataset1
2 ls
3
4 # FASTQ paired end read files
5 >ctrl1.fastq.gz ctrl2.fastq.gz ctrl3.fastq.gz
6 >test1.fastq.gz test2.fastq.gz test3.fastq
7
8 # Use {zless or zcat} if your data is gzipped.
9 zless ctrl1.fastq.gz # OR
10 zcat ctrl1.fastq.gz | head
11
12 # OR
13
14 # Use {less or head or more} if your data is text.
15 head test3.fastq # OR
16 more test3.fastq # OR
17 less test3.fastq # Remember: less is more ;))
18
19 @SRR1660397.66 HWI-ST992:140:C2134ACXX:1:1101:5890:2171 length=50
20 GTTCAAAGATGAACTAGAAGACAGAAATACTGTTCAGGAGTATGGCTGTC
21 +
22 @@@DDDDDHHHHHGGGGIDHGCBHIIIFHIHIH?GHIJFG?DFGGIJGFG
The general structure of the tophat command is:
1 tophat2 [options] <bowtie_index>
2 <reads1[,reads2,...]> [reads1[,reads2,...]] \
3 [quals1,[quals2,...]] [quals1[,quals2,...]]
- It is common to give tophat a transciptome index, .GTF file with annotations but not necessary for small sets.
- Most of the option/parameters in tophat have been optimized for standard runs.
NOTE: Since we will use several options and since these options make typing out the command every time a problem we will produce a small script so that we may run it in the bash shell.
- Go to the command line:
touch tophat2.sh - Open nano a text editor:
nano tophat2.sh - In nano text editor, type:
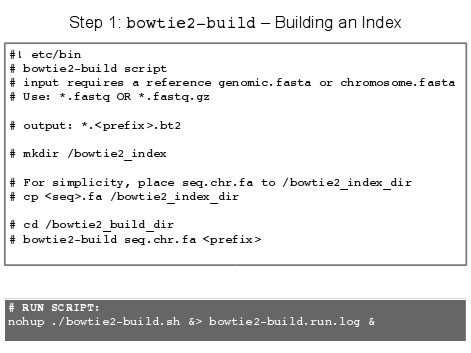
IMPORTANT: Tophat requires a bowtie2 index tto speed alignment, which can be generated by bowtie-build
- Save the above batch file into the appropriate directory.
- Now, make the script file executable:
chmod +x tophat2.sh - To run this batch file, goto command line: nohup ./tophat.sh >& tophat.run.date.log &
Nohup
- Nohup: Run a Command or Shell-Script Even after You Logout Tophat may take some time to run depending on the size of the read files from the RNA-seq experiment.
- With nohup
command>&mylogfile & before and after the command, you are running the software in background, and come back to check the process later. - If you need to terminate a program that is NOT run in the background, press “Ctrl-C” to stop it.
- If you need to terminate a program that is run in the background, first use “top” or “ps -u my_user_ID” to find out the process ID (PID) of your program, it should be an integer number. Then use the command “kill PID” to terminate the program. Make sure you use “top” again to confirm that the program is actually killed.
- When you use a shell script to run a batch of commands, you will need to use “kill PID” to kill both the PID of the shell script, and the any of the commands that are still running.

- Once the tophat has finished, we can inspect the results.
- In our results directory, we should find 8 files & 1 directory (logs).
- accepted_hit.bam
- accepted_hit.bam.bai
- align_summary.txt
- deletions.bed
- insertions.bed
- junctions.bed
- prep_reads.info
- unmapped.bam
- DIR /logs
- To view .BAM files we can use samtools view
- The .BAM file is a binary alignment mapping file.
- If you try to open this file in a normal text editor you will see nonething intelligble.
- SamTools can read binary files
- Type: samtools view accpeted_hits.bam
The deletion, insertion and junction .bed files do not tell me much, :))
- View *.bed files,
- Type: less deletions.bed
- Type: less insertions.bed
- Type: less junctions.bed
The align_summary.txt is filled with interesting information.
- There is align_summary.txt which can be very useful to inspect.
- Type: nano align_summary.txt
The align_summary.txt file will have information regarding the number of reads mapped. For example, you may have left reads, right reads and information on the concordant and discordant alignments.
Q. What are concordant and discordant alignments?
Contents of align_summary.txt:
1 Left reads:
2 Input : 60586968
3 Mapped : 58163843 (96.0% of input)
4 of these: 6832240 (11.7%) have multiple alignments (359075 have >10)\
5
6 Right reads:
7 Input : 60586968
8 Mapped : 56969290 (94.0% of input)
9 of these: 6668479 (11.7%) have multiple alignments (359075 have >10)\
10
11 95.0% overall read mapping rate.
12
13 Aligned pairs: 55880048
14 of these: 6491876 (11.6%) have multiple alignments
15 2795712 ( 5.0%) are discordant alignments
16 87.6% concordant pair alignment rate.
For more information on the subject, see Biostars: “Question: Tophat2 align_summary interpretation”
- Once we have run Tophat2 and received our data, we can use grep for more information retrieval.
- Since we have multiple test and cpntrol file to investigate we can: **cd ~/parent_dir
- In your shell type: **grep “overll read mapping rate” */align_summary.txt **
- Find the overall rad mapping rates for the different samples in our experiment.
1 95.0% overall read mapping rate.
2 93.0% overall read mapping rate.
3 98.0% overall read mapping rate.
4 95.2% overall read mapping rate.









5.X - Papers on the Topic of RNA-Seq
- Computational methods for transcriptome annotation and quantification using RNA-seq, Manuel Garber, et al, Nature Methods 8, 469–477 (2011)
- From RNA-seq reads to differential expression results, Alicia Oshlack, et al, Genome Biology, 2010, 11:220
- RNA-Seq: a revolutionary tool for transcriptomics, Zhong Wang, Nature Reviews Genetics 10, 57-63 (January 2009)
- RNA sequencing: advances, challenges and opportunities, Fatih Ozsolak, et al, Nature Reviews Genetics 12, 87-98 (February 2011)
- RNA-Seq—quantitative measurement of expression through massively parallel RNA-sequencing, Brian T. Wilhelm, et al, Methods, Volume 48, Issue 3, July 2009, Pages 249-257
- The simple fool’s guide to population genomics via RNA-Seq: an introduction to high-throughput sequencing data analysis, De Wit P, et al, Mol. Ecol. Resour. 2012 Nov; 12(6): 1058-67
- Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks, Cole Trapnell, et al, Nature Protocols, 7, 562–578, (2012)
- Next-generation transcriptome assembly, Zhong Wang, et al Nature Reviews Genetics 12, 671-682 (October 2011)
- Informatics for RNA Sequencing: A Web Resource for Analysis on the Cloud, Malachi Griffith, et al, PLOS, Published: August 6, 2015, https://doi.org/10.1371/journal.pcbi.1004393
- A survey of best practices for RNA-seq data analysis, Ana Conesa, et al, Genome Biology 2016 17:13
- Computation for ChIP-seq and RNA-seq studies, Shirley Pepke, et al, Nature Methods 6, S22 - S32 (2009)
- Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-seq experiments, N. Vijay, et al, Mol. Eco., Volume 22, Issue 3, February 2013, Pages 620–634
- RNA-Seq Assembly – Are We There Yet?, Simon Schliesky, et al, Front Plant Sci. 2012; 3: 220
Web sites with additional information:
- https://www.genome.gov/13014330/
- http://www.cd-genomics.com/RNA-Seq-Transcriptome.html
- https://www.rna-seqblog.com/rna-seq-a-tutorial/
- https://www.biostars.org/p/52152/
Genes and disease: Encore une fois, The genomic era arrives. And this time it’s probably real “Encore une fois” - Once again
https://samtools.github.io/hts-specs/SAMv1.pdf
http://www.htslib.org/doc/samtools.html